臨床検査サービス | 新着情報
- 2025/12/04
- CTCラボのインタビュー記事がバイオ・ラッド社HPに掲載されました
- 2025/8/22
- 動物iPS細胞・ES細胞 Gバンド染色体解析サービスのご案内
- 2025/7/31
- CTCラボニュース 新たな展開(2025年8月より)
- 2024/11/14
- 韓国 臨床統合医学癌学会 第15回秋季国際学術セミナーにおいて講演いたしました
多発性骨髄腫,精神発達遅滞,ファンコニ貧血,マルファン症候群,リンチ症候群,染色体数的異常,加齢黄斑変性症,カウデン病
■MM(多発性骨髄腫)とは
多発性骨髄腫(MM)は、骨髄に蓄積する形質細胞の悪性腫瘍であり、骨破壊および骨髄機能不全にかかわる。多発 性骨髄腫は形態学的に類似しているが、疾患が持ついくつかのサブタイプは、遺伝子および分子レベルで識別できる。初期診断における骨髄検査には、染色体分析やおよびFISHによる解析により、転座、欠失、または増幅により染色体 異常が識別されています。
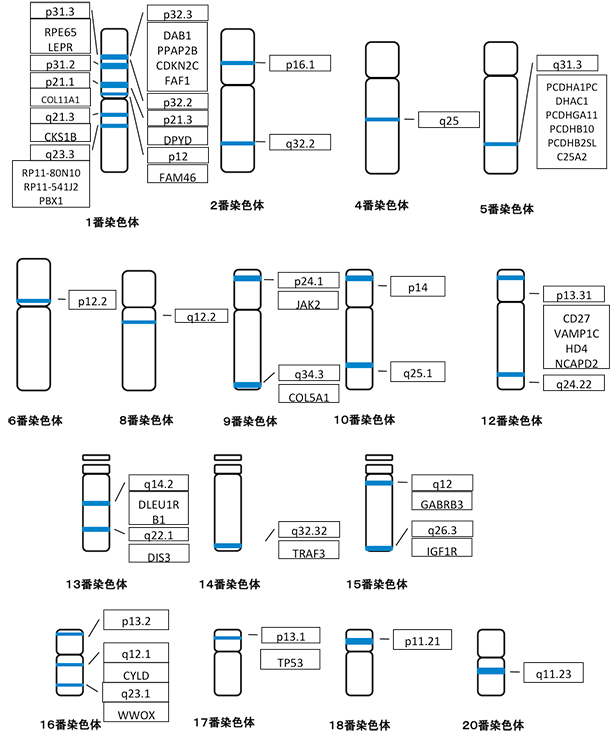
13番染色体 del(13) の欠失は、細胞周期の遺伝子発現に増幅効果をもたらすとされており、総合的に生存率にかかわると言われています。染色体17番p13に存在するp53の欠失は、TP53のヘテロ接合体の損失につながり、MMではハイリスク状態とみなされる。また、染色体14番q32に位置するIgh遺伝子がかかわる染色体異常として、t(4;14)(p16;q32) t(11;14)(q13;q32) t(14;16)(q32;q23) t(14;20)(q32;q11.2) などの転座がみられ、t(4;14)は全体の15%前後に認める重要な転座とされています。
転座のほか、1番染色体短腕の欠失、長腕の増幅も多く報告され、予後不良因子とされており、1q21の増幅はMMへの進展に関係すると示唆されています。
■日本遺伝子研究所で行っている主な事例
・ MRC-Holland社のMLPA kit を使用して染色体検査では検出できない染色体異常(微細欠失)を検出することができます。
・MMで出現頻度の高い、13番染色体の欠失(RB1)、17番染色体(TP53)を同時に解析できます。
・1p欠失、1qの重複など、予後不良因子で見られる異常を中心に解析する構成になっています。
・染色体が得られない場合でも解析できます。
・同時に多項目の遺伝子の欠失を検出することができます。
■日本遺伝子研究所だからできる特徴
MLPA法の解析では14番染色体の融合遺伝子を検出する設定がないため、染色体検査やFISH検査と組み合わせて解析することで、より精度の高い解析結果を得ることができます。
■MR(精神発達遅滞)とは
全般的な知能の発達に遅れがみられ、社会生活にうまく適応できない状態です。生まれたときからほぼ18歳くらいまで(発達期)に低い知能がみられる場合をいいます。脳になんらかの原因があって、精神遅滞をきたす場合、障害の程度の重いことが多くみられ、原因となる病気はさまざまです。
原因不明を呈する発達遅延および/または同時に複数の微小欠失症候群の精神遅滞の患者をスクリーニングするために開発されたMLPAプローブを使用します。
対象疾患
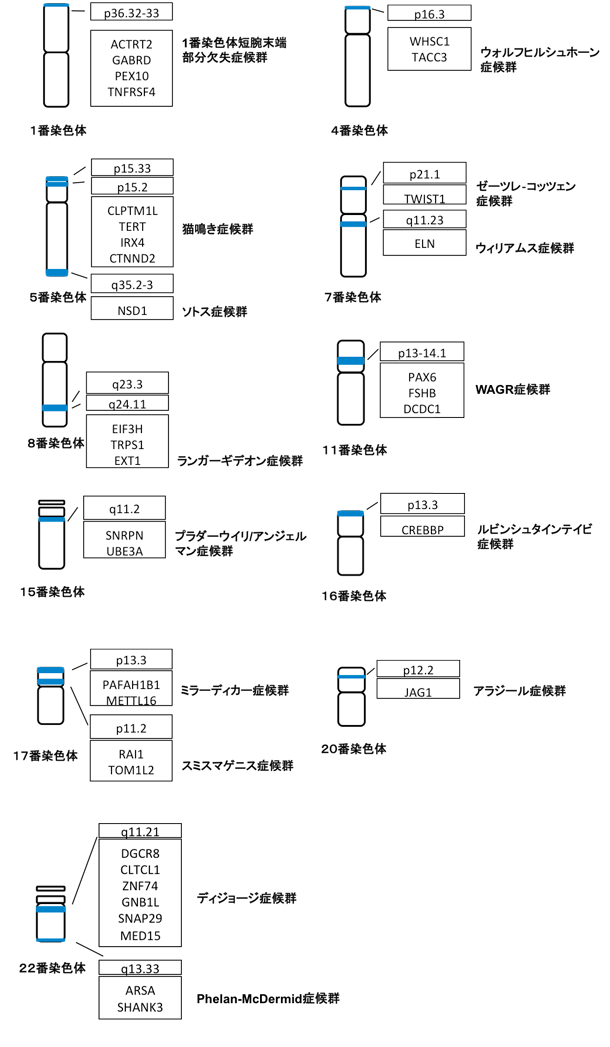
・1番染色体短腕末端部分欠失症候群
・猫鳴き症候群
・ゼーツレ‐コッツェン症候群
・ランガーギデオン症候群
・プラダーウイリ/アンジェルマン症候群
・ミラーディカー症候群
・アラジール症候群
・Phelan-McDermid症候群
・ウォルフヒルシュホーン症候群
・ソトス症候群
・ウィリアムス症候群
・WAGR症候群
・ルビンシュタインテイビ症候群
・スミスマゲニス症候群
・ディジョージ症候群
■日本遺伝子研究所で行っている主な事例
・MRC-Holland社のMLPA kit を使用して染色体検査では検出できない染色体異常(微細欠失)を検出することができます。
・同時に多項目の遺伝子の欠失を検出することができます。
■日本遺伝子研究所だからできる特徴
FISH法では多項目を解析するのに複数種類のFISHプローブを必要としますが、MLPA法の遺伝子解析を実施することで、一度に多項目の原因の特定されている精神遅滞を解析できます。
■FANCA/FANCB/FANCD2/PALB2/RAD50/RAD51C遺伝子(ファンコニ貧血) とは
ファンコニ貧血(FA)の患者はマイトマイシンCやDEBなどのDNA二重鎖架橋形成物質に感受性が高く、末梢血リンパ球培養時にこれらを添加することで染色体の切断・染色分体交換の数が増えることから鑑別のために実施されます。
汎血球減少、皮膚の色素沈着、奇形、低身長、性腺機能不全をともなう、常染色体の劣性疾患です。FAは遺伝的に多様な疾患であり、現在までに13の責任遺伝子が同定されています。
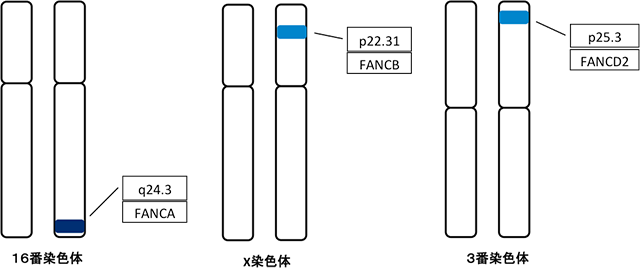
その中で、染色体16q24.3の上のFANCA遺伝子の何らかの変異は約56%で見られ、ホットスポットと言えるような特徴的な変異は見られず、ゲノム単位の広範な欠失の頻度が高いことが報告されています。
FAに含まれる遺伝子はすべてDNA修復に関係しており、染色体切断はFAの特徴で、その染色体不安定性を背景に、進行性汎血球減少、MDSや白血病への移行、身体奇形、固型癌の合併を特徴とする血液疾患です。
日本での年間発生数は5〜10人で、出生100万人あたり5人前後で、海外からの報告とほぼ同程度とされています。常染色体劣性の遺伝形式をとることから、そのキャリア頻度は、200〜300人に1人と推定されています。
■日本遺伝子研究所で行っている主な事例
・ MRC-Holland社のMLPA kit を使用して染色体検査では検出できない染色体異常(微細欠失)を検出することができます。
・染色体が得られない場合でも解析できます。
・FANCA遺伝子では16番染色体のFANCA遺伝子のエクソン領域を特異的に検出するプローブを用いて解析します。
・X染色体のFANCB遺伝子のエクソン領域を特異的に検出するプローブを用いて解析します。
・3染色体のFANCD2遺伝子のエクソン領域を特異的に検出するプローブを用いて解析します。
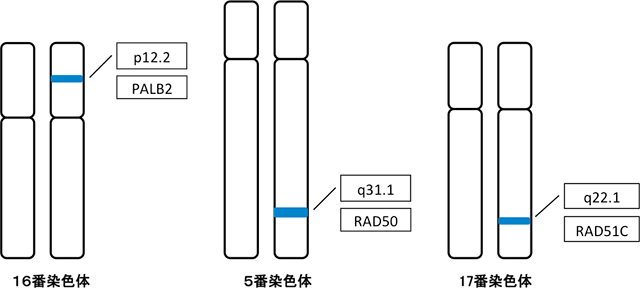
・16染色体のPALB2遺伝子のエクソン領域を特異的に検出するプローブを用いて解析します。
・5染色体のRAD50遺伝子のエクソン領域を特異的に検出するプローブを用いて解析します。
・17染色体のRAD51C遺伝子のエクソン領域を特異的に検出するプローブを用いて解析します。
■日本遺伝子研究所だからできる特徴
マイトマイシンCやDEBを添加した染色体脆弱試験の実施の他、MLPA法による遺伝子解析を同時におこなうことにより、より精度の高い解析結果を得ることができます。
ファンコニ貧血の解析として
FANCA遺伝子
FANCB遺伝子
FANCD2/ PALB2遺伝子
PALB2/RAD50/RAD51遺伝子
の解析項目があります。
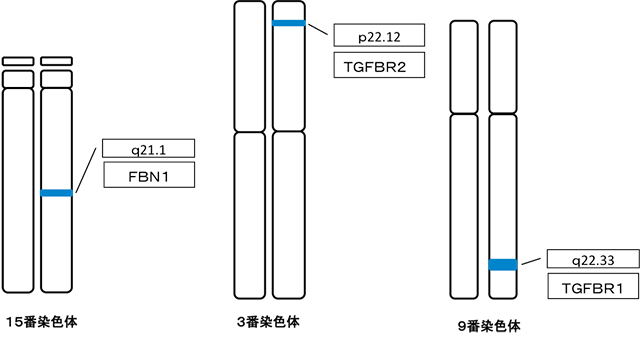
■ FBN1/TGFBR1/TGFBR2遺伝子(マルファン)症候群 とは
マルファン症候群は、線維性結合組織の遺伝性疾患です。結合組織は全身に存在するため、大動脈瘤や大動脈解離、高身長、側弯等の骨格変異、水晶体亜脱臼、自然気胸など様々な症状を呈し、生まれたときから非常に重 篤な症状もあれば中高年になるまで大きな症状が出ることがない、あるいは循環器系の症状が重い、または知的障害を併せ持つ症状、など様々です。 発生頻度は全ての人種と男女にかかわらず3,000〜10,000人あたり1人といわれ、日本には20,000人いると推定されます。
現在、1型、2型が認められており、前者は、15番染色体上のフィブリリン1 (fibrillin-1, FBN1) の先天性遺伝子異常、後者は3番染色体上のがん抑制遺伝子TGFBR2の先天性遺伝子異常が原因で共に常染色体優性遺伝により50%の確率で子供に遺伝する可能性があります。また、TGFBR1にも変異が見つかった例が報告されています。
本解析では、MLPA法を用いFBN1遺伝子またはTGFBR1,TGFBR2遺伝子におけるエクソンレベルの比較的大きな重複や欠失を解析します。
■日本遺伝子研究所で行っている主な事例
・ MRC-Holland社のMLPA kit を使用して染色体検査では検出できない染色体異常(微細欠失)を検出することができます。
・染色体では10bp以上の大きい欠失でないと検出できないので、エクソン領域で欠失が分かるMLPA法の遺伝子解析でなければ解析できません。
・ FBN1遺伝子では、エクソン1〜66までの領域を特異的に検出するプローブを用いて解析します。
・ 9番染色体のTGFBR1遺伝子のエクソン1〜9までの領域を特異的に検出するプローブを用いて解析します。
・TGFBR2遺伝子では3番染色体のエクソン1〜8までの領域を特異的に検出するプローブを用いて解析します。
■日本遺伝子研究所だからできる特徴
エクソン領域を細かく解析するので、どのエクソン領域の欠失が原因か解析することができます。
マルファン症候群の解析として
FBN1遺伝子
TGFBR1/TGFBR2遺伝子
の解析項目があります。
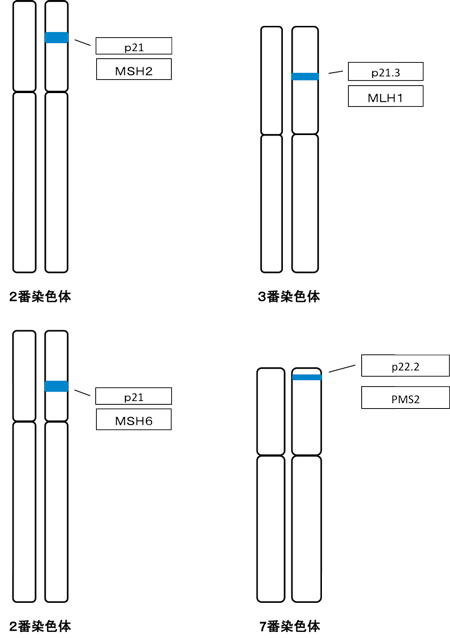
■ MSH2/ MLH1/ MSH6/ PMS2遺伝子(リンチ症候群) とは
リンチ症候群(遺伝性非ポリポーシス性大腸がん:Hereditary Non-Polyposis Colorectal Cancer:HNPCC)は、家族性大腸腺腫症(Familial Adenomatous Popyposis : FAP)とともに遺伝性の大腸がんの中で頻度が高いものとして知られています。
家族性大腸腺腫症は、通常大腸に100個以上の多発性腺腫が見られるため、臨床的に診断されやすい常染色体優性遺伝性の腫瘍です。
一方、リンチ症候群は、大腸がんや子宮内膜がんを高率に発症する特徴があります。ミスマッチ修復機構に関係する4つの遺伝子(MSH2, MLH1, MSH6, PMS2)の片方のアレルに変異を持ち(キャリア)、もう片方に後天的に変異が加わることによりミスマッチ修復機構が損なわれ腫瘍が発生すると考えられています。
全大腸がんの1〜5%を占めると推定されています。
■日本遺伝子研究所で行っている主な事例
・MRC-Holland社のMLPA kit を使用して、MSH2, MLH1, MSH6, PMS2 の4つの遺伝子についてエクソン単位の欠失や増幅を解析します。
・これらの4つの遺伝子は、リンチ症候群の原因遺伝子として現時点で98%以上を占めるといわれています。
■日本遺伝子研究所だからできる特徴
エクソン領域を細かく解析するので、どのエクソン領域の欠失が原因か解析することができます。
リンチ症候群の解析として
MLH1/MSH2/BRAF/CDKN2A遺伝子
MSH6/MLH1/MSH2/MUTYH遺伝子
PMS2/PMS2CL遺伝子
の解析項目があります。
■ Aneuploidy(染色体数的異常)とは
ヒトの染色体数は通常46本(2n=46)ですが、染色体数的異常は、22種類の常染色体と2種類の性染色体のどれ かが一つ以上増減します。ほとんどは、染色体が1本多いトリソミーか1本少ないモノソミーであり、よくみられる染色体数的異常には、21トリソミー(ダウン症候群)、18トリソミー(エドワード症候群)、13トリソミー(パトー症候群)、クラインフェルター症候群(47,XXY)、ターナー症候群(45,X)があります。
ダウン症候群は最も一般的な先天性疾患の一つで、約800人に1人の割合で出生すると言われています。ダウン症候群は21番染色体の遺伝子の過剰なコピー数の存在により特徴付けられるとされ、ダウン症候群の約95%は21トリソミーが原因とされています。また、1-2%の割合で21番染色体のモザイクがダウン症候群で見られます。21番染色体の一部分の重複は、部分的もしくは完全型としてダウン症候群の表現型が現れると言われています。家族性のダウン症候群として、ロバートソン型転座のダウン症候群があり、全体の1-2%に見られます。
エドワーズ症候群はダウン症候群の次に一般的な常染色体性トリソミーです。約6000人に1人の割合で発生するとの報告があります。
また、13トリソミーは、10000人に1人の割合で新生児に発生すると言われており、18トリソミーと13トリソミーを持つ多くの胎児は妊娠初期で流産してしまうことが報告されています。
性染色体に関する症候群は、13番、18番、21番のトリソミーの頻度よりも高く、X、Y染色体の異数性は、様々な障害が発生することが知られています。
■日本遺伝子研究所で行っている主な事例
本解析では、MRC-Holland社のMLPA kit を使用して、ゲノムDNAから13番染色体、18番染色体、21番染色体、X染色体、Y染色体の増減を解析します。

■日本遺伝子研究所だからできる特徴
弊社は、MLPA法に先んじてG-band法とFISH法による染色体数的異常の解析を実施してまいりました。
G-band法は、細胞が分裂していないと染色体が得られないため解析することができませんが、上述の5種類の染色 体に限定されず、全染色体の数的変化および構造的な変化を数Mbpレベルで確認することができます。
FISH法は、一度に解析できる領域の数が限られ、Aneuploidyにおいては13、18、21、X、Y染色体上の各々一領域のみですが、細胞が分裂していなくても解析することが可能な上、核酸抽出の手間が不要で3法の中では比較的迅速に、数10kbp〜数Mbpレベルの変化を確認することができます。
MLPA法( MRC-Holland社のMLPA kit)は、FISH法で各々一領域の検出に限られていた変化を13、18、21、X染色体上で各8箇所ずつ、Y染色体上で4箇所検出できますのでG-band法とFISH法で検出できない部分欠失や増幅を捉えることが可能です。
状況に合わせて3種類の解析法を使い分ける若しくは、併用することができますので効率よく数的異常を解析することができます。
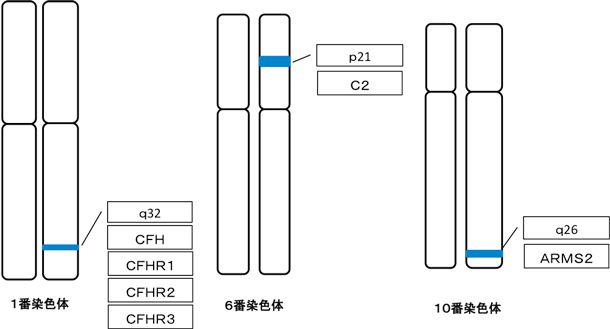
■ ARMD(加齢黄斑変性症) とは
加齢性黄斑変性症(ARMD)は、高齢者の失明の主な原因です。中心視野視力喪失を引き起こし、網膜の中心領域(黄斑)の進行性破壊を特徴とします。環境要因と遺伝的感受性の影響が確認されています。
加齢性黄斑変性の遺伝的寄与リスクの約50%は、1番染色体長腕部のCFH遺伝子領域におけるコピー数の変化との関係が報告されています。CFH遺伝子は1q32に位置して、23個のエクソンから構成され、97.7kbのサイズがあります。2番目の原因遺伝子は、10番染色体長腕部10q26上のARMS2遺伝子(LOC387715)と報告されています(SNP rs10490924) 。3番目は、C2/CFB領域のSNP(rs9332739及びrs641153)が報告されています。 C2およびCFBの遺伝子は染色体6p21上、補体成分2および補体因子Bをコードしています。
■日本遺伝子研究所で行っている主な事例
・MRC-Holland社のMLPA kit を使用して、ゲノムDNAから1番染色体上のCFH、CFHR1 、CFHR2 、CFHR3 、 CFHR5遺伝子、6番染色体上のC2遺伝子および10番染色体上のARMS2遺伝子のエクソンレベルのコピー数の変化を解析します。
■日本遺伝子研究所だからできる特徴
臨床検体としての加齢黄斑変性症の解析の他、研究解析で使用される培養細胞においても解析材料として使用することができます。
■ PTEN(カウデン病) とは
カウデン病は多発性過誤腫症候群とも呼ばれ、歯肉に乳頭腫様の病変や手足に小さな角化性の丘疹、顔面に外毛根鞘腫などの良性の角化性皮膚腫瘍が複数発生します。また、食道から大腸におよぶ消化管ポリポーシスを生じます。乳房、子宮、甲状腺などいろいろな臓器に悪性腫瘍ができることもあります。
常染色体優性遺伝形式をとることが知られており、80%の割合でPTEN遺伝子に生まれ持った塩基配列の変異があるという報告がされています。これらは、エクソン領域のフルシークエンス解析で検出することができますが、それでも、変異が検出できなかった場合、MLPA法によってエクソンの欠失や増幅の有無を確認します。
■日本遺伝子研究所で行っている主な事例
・MRC-Holland社のMLPA kit を使用して、ゲノムDNAよりPTEN遺伝子のエクソンレベルの比較的大きな欠失や重複を解析します。
■日本遺伝子研究所だからできる特徴
エクソン領域を細かく解析するので、どのエクソン領域の欠失が原因か解析することができます。
■お問い合わせ
(株)日本遺伝子研究所 検査事業部
TEL:022-388-9747 FAX:022-388-9740
お問い合わせフォームはこちら

